Helmsman: fast and efficient mutation signature analysis for massive sequencing datasets

![]()
The spectrum of somatic single-nucleotide variants in cancer genomes often reflects the signatures of multiple distinct mutational processes, which can provide clinically actionable insights into cancer etiology. Existing software tools for identifying and evaluating these mutational signatures do not scale to analyze large datasets containing thousands of individuals or millions of variants.
We introduce Helmsman, a program designed to perform mutation signature analysis on arbitrarily large sequencing datasets. Helmsman is up to 300 times faster than existing software. Helmsman’s memory usage is independent of the number of variants, resulting in a small enough memory footprint to analyze datasets that would otherwise exceed the memory limitations of other programs.
Helmsman is a computationally efficient tool that enables users to evaluate mutational signatures in massive sequencing datasets that are otherwise intractable with existing software. Helmsman is freely available at https://github.com/carjed/helmsman.
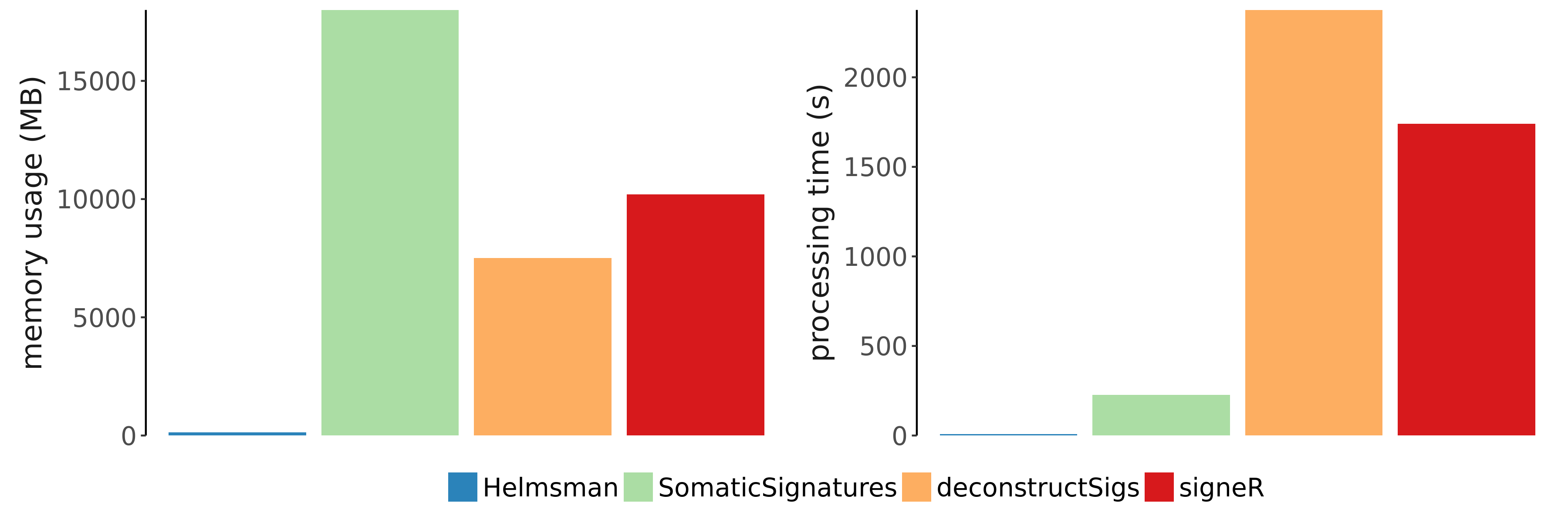
Figure 1
Helmsman is orders of magnitude faster and more memory efficient than competing mutation signature analysis tools
Citation
If you use Helmsman in your research, please cite the following publication:
- Carlson, J, Li, JZ, Zöllner, S. Helmsman: fast and efficient mutation signature analysis for massive sequencing datasets. BMC Genomics. 2018;19: 845.
doi:10.1186/s12864-018-5264-y